
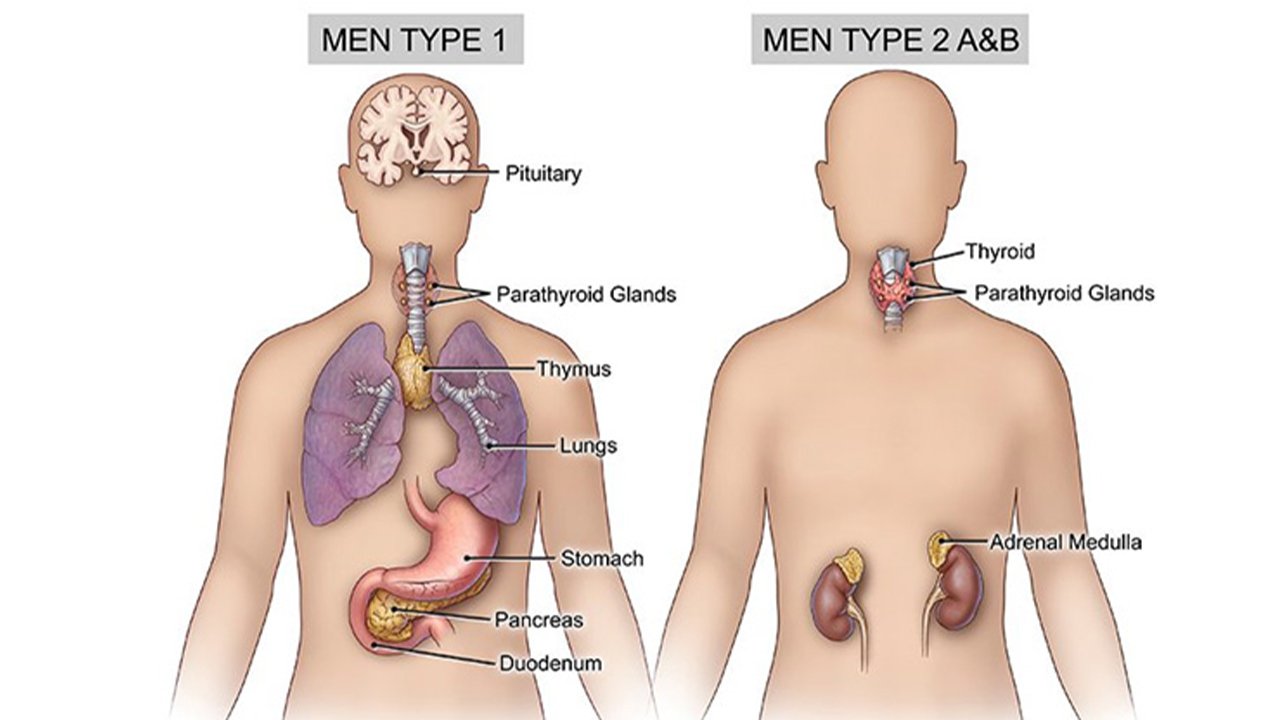
What is Multiple Endocrine Neoplasia-1 (MEN-1) Syndrome?
MEN1 gene is located on chromosome 11q13 and encodes the protein menin. Majority of mutations that cause MEN-1 syndrome are nonsense or frameshift. It is an Autosomal Dominant disorder that predisposes an individual to following malignancies-
Parathyroid tumors and Primary Hyperparathyroidism
Most common features and often the first presenting sign of MEN1. It usually presents as severe hypercalcemia with symptoms such as “stones, bones, abdominal groans, psychic moans”. Sometimes, it may also cause hypertension, or shortened QT interval on ECG.
Duodenopancreatic Neuroendocrine Tumors
Most common are gastrinomas, that are multicentric, small (<0.5cm) foci, throughout the duodenum. Most result in peptic ulcer disease (Zollinger-Ellison syndrome) and 50% are malignant. Others include Insulinomas, VIPomas, Glucagonomas, and Somatostatinomas.
Pituitary tumors
Most commonly seen are microadenomas (<1 cm) and prolactinomas.
Two of these three major syndromes must be present to be classified as MEN-1. When it is present in 1 more first degree relative, it is called as Familial MEN-1 syndrome.
How is the surveillance or screening done in MEN-1 syndrome?
Pituitary Tumors– Serum prolactin and/or IGF-1 monitoring to be started at 5 years of age and to be done annually. Brain MRI to be done starting at 5 years and then every 3-5 years based on biochemical results.
Parathyroid Tumors And PHPT – Fasting total serum calcium and/or ionised calcium and PTH starting at 8 years of age and tp be checked every year.
Duodenopancreatic Gastrinoma – Fasting serum gastrin monitoring starting at 20 years of age and to be done annually.
Pancreatic NET – Chromogranin A, PP, VIP, glucagon levels to be checked yearly starting before 10 years of age. Abdominal CT and MRI starting at 20 years of age and every 3-5 years based on biochemical results.
Insulinoma – Fasting glucose and insulin to be checked every year starting at 5 years of age.
What are the therapeutic interventions that may be required for MEN-1 syndrome?
Parathyroid Tumors
- Subtotal parathyroidectomy (removal of 3-3.5 glands)
- Total parathyroidectomy with autotransplantation of parathyroid tissue into the forearm
- Management of persistent hypoparathyroidism with oral calcitriol
Duodenopancreatic NET: Surgical resection of the tumor
Pituitary Tumors: They are usually treated with medical therapy
What is Multiple Endocrine Neoplasia-2 (MEN-2) Syndrome?
It is an autosomal dominant disorder caused by inherited mutations in the RET gene, located on Chromosome 10q11.2, rearranged during Transfection. It encodes a receptor tyrosine kinase with extracellular, transmembrane and intracellular domains.
Table below describes the various types of MEN-2 syndromes and their characteristic malignancies-
|
Subtype |
Medullary Thyroid Ca (%) |
Pheochromocytoma (%) |
Parathyroid Disease (%) |
|
MEN2A |
95 |
50 |
15-20 |
|
FMTC |
100 |
0 |
0 |
|
MEN2B |
100 |
50 |
Uncommon |
Other lesions that may be seen are as follows-
MEN2A
- cutaneous lichen amyloidosis
MEN2B
- Diffuse ganglioneuromas of GI tract
- Mucosal neuromas over lips, tongue, palate, pharynx
- Marfanoid habitus
- Prominent thickened corneal nerves
Hereditary Medullary Thyroid Carcinoma (MTC)
It is commonly the first manifestation of MEN-2 syndrome, with 20-25% being hereditary. If not screened, it presents with a neck mass at 5-20 years of age. It is mostly bilateral and/or multifocal and arises in a background of C-cell hyperplasia. Biochemical manifestations usually appear between 5 and 25 years of age. Most frequent first symptom is diarrhea. In patients with plasma calcitonin >10 ng/ml, the disease has poor prognosis.
Pheochrocytoma
MEN-2 usually presents with early onset (<35y), bilateral pheochromocytomas, with personal and/or family history of MTC or heperparathyroidism. Most common presenting feature is intractable hypertension or anesthesia induced hypertensive crisis.
Primary hyperparathyroidism
It is usually multiglandular and presents earlier (Before 6th decade). It may be in the form of adenoma or glandular hyperplasia parathyroid glands.
What are the therapeutic interventions that may be required for MEN-1 syndrome?
Risk reducing thyroidectomy
Risk-reducing thyroidectomy and parathyroidectomy with reimplantation of one or more parathyroid gland into the neck or non dominant forearm.
Recommendations for screening and prophylactic thyroidectomy based on risk-
|
Risk Level |
Age Of RET Testing |
Timing Of Prophylactic Thyroidectomy |
|
D |
ASAP And With First Year Of Life |
ASAP And Within First Year Of Life |
|
C |
<3-5 Y |
Before Age 5 Y |
|
B |
<3-5y |
Consider Surgery Before Age Of 5y. May Delay If Criteria Are Met |
|
A |
<3-5 Y |
Screening at-risk individuals for pheochromocytoma/ hyperparathyroidism
ATA recommends annual screening after 8 years of age for those with RET mutations in codon 630 and 634 and RET mutation associated with MEN2B. In the presence of other MEN2A RET mutations, annual screening is recommended by age 20 years.
Biochemical tests used for screening are plasma-free metanephrines, and/or urinary fractionated metanephrines. MRI is done if biochemical tests are abnormal.
Treatment of MTC/ Pheochromocytoma/ Hyperparathyroidism
Treatment Of MTC:
Standard treatment in case of adults is surgical removal of entire thyroid gland with posterior capsule and central lymph node dissection, followed by lifelong thyroid hormone replacement therapy. Whereas, in adults with metastatic disease who are ineligible for surgery, Vandetanib may be given.
Treatment Of Pheochromocytoma:
In case of unilateral tumors, laparoscopic adrenalectomy is done, whereas, Cortical-sparing surgery is done for bilateral ones. It minimizes the risk of adrenal insufficiency and the need for lifelong steroid supplementation. Following this, surveillance is required for new or recurrent disease.

